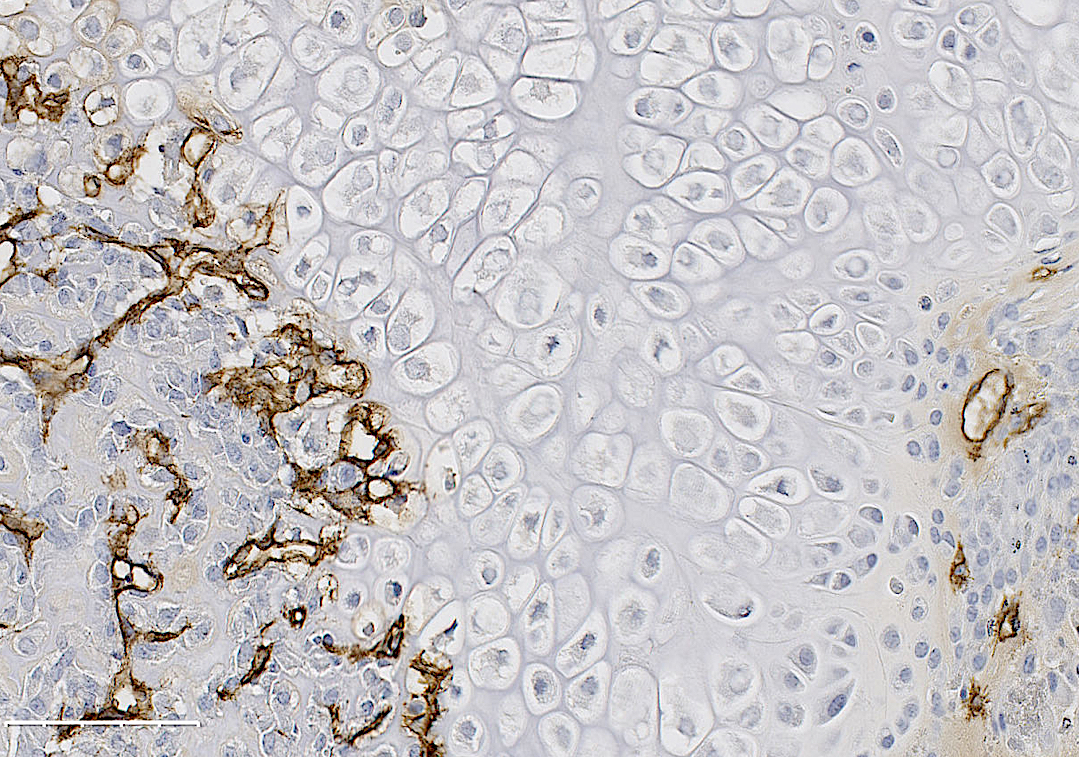
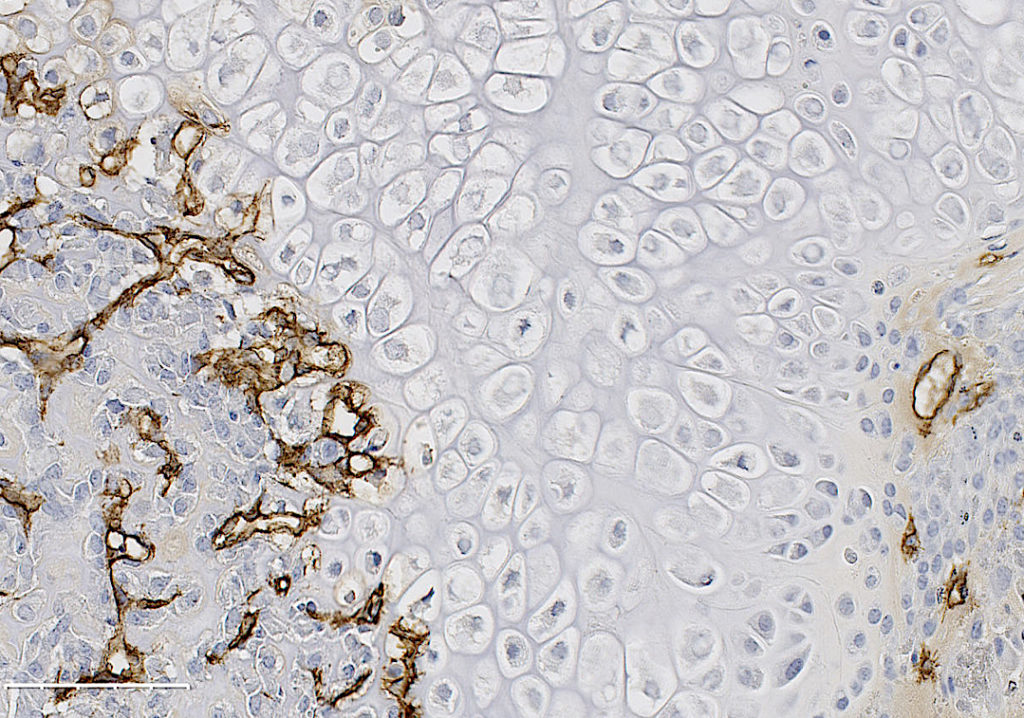
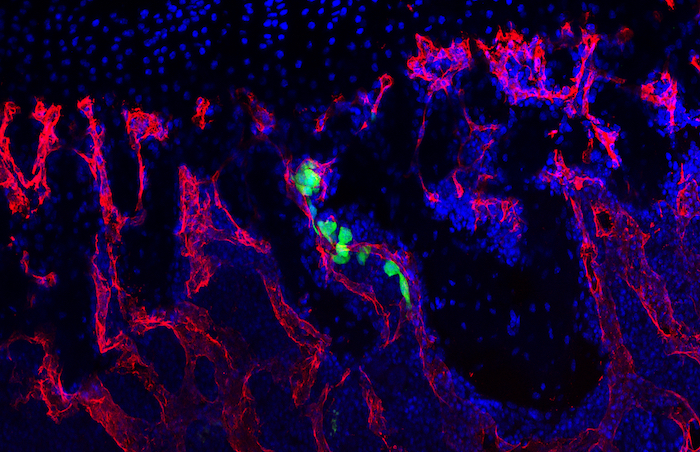
Formation of new blood vessels is necessary for proper bone fracture healing. In support of this, fracture patients with pre-existing conditions associated with vascular disease and injury, such as smoking, advanced age, diabetes or other large-scale soft tissue trauma have lower successful fracture healing rates. Vascular endothelial growth factor A, abbreviated as VEGFA, is essential in coordinating the proliferation and recruitment of existing blood vessels across a range of bodily injury sites. In bone, blocking VEGFA with certain pharmacological agents, has been shown to inhibit bone fracture healing. However, since these pharmacological approaches are systemic and nonspecific, the cells in bone that make VEGFA and are necessary for blood vessel formation during fracture healing, remain poorly defined. This work is important to understanding why certain fractures won’t heal and would inform future treatment strategies.
Recent work in the role of blood vessels in bone came from Henry Kronenberg and Crista Maes at Harvard. These authors demonstrated that VEGFA expressing Osterix (Osx) lineage cells, a marker of early osteoprogenitors which give rise to bone forming cells called osteoblasts, secrete VEGFA and invade the fracture callus with blood vessels. Furthermore, the team of Bjorn Olsen at Harvard, demonstrated via genetic deletion of VEGFA from Osx lineage cells that blood vessel formation during cortical defect healing was impaired. However, this localized bone injury doesn’t recapitulate all aspects of fracture repair, including periosteal activation in the outer lining of the bone. Building upon these important works, our lab group at WUSTL decided to test the role of VEGFA from osteoprogenitors (Osx lineage) and their more mature cells osteoblasts and osteocytes, marked by Dentin Matrix Protein 1 (DMP1), during clinical fracture repair models such as stress fractures and full fracture.
In our study, published in JBMR in 2019, we performed genetic inducible deletion of VEGFA in different cell types. We deleted VEGFA from all cells, or from bone-forming osteoblast cells at 2 stages of maturation; osteoprogenitors and mature osteoblasts/osteocytes. We then performed either a full semi-stabilized fracture or stress fracture. Cortical defect healing was also tested to compare to the results of the Olsen team. Our study revealed that loss of VEGFA in all cells blocked bone and blood vessel formation regardless of the injury model, reaffirming previous pharmacological results. More importantly, we showed that VEGFA from osteoprogenitor cells (Osx lineage) but not more mature osteoblasts/osteocytes (DMP1 lineage) was necessary for maximal bone formation and blood vessel formation in stress fracture and full fracture models. Surprisingly, VEGFA from osteoprogenitor cells at the time of injury was not important for cortical defect repair. These results together, highlight that the VEGFA cell source necessary for bone healing depends on the type of bone injury. Furthermore, our study is the first to indicate that VEGFA from different osteoblast maturation stages play nonredundant roles during clinical models of fracture healing. Our results may also help explain why recombinant BMPs, which stimulate VEGFA production in osteoprogenitors, are promising treatments to induce bone formation during fracture. Overall, these findings highlight that osteoprogenitors are critically sources of VEGFA during fracture healing. Furthermore, these same cells should be clinical targets for improving bone blood vessel formation to promote fracture repair in patients with poor healing outcomes.
These findings are described in more detail in the article entitled “VEGFA From Early Osteoblast Lineage Cells (Osterix+) Is Required in Mice for Fracture Healing” published in JBMR. The work was conducted by Evan Buettmann, Jennifer McKenzie, Nicole Migotsky, David Sykes, Pei Hu, Susumu Yoneda, Matthew Silva. This work was funded by NIAMS (R01 AR050211 and P30 AR057235). The authors would like to thank the Washington University in St. Louis Musculoskeletal Research Center (MRC) Cores and staff for histological and micro-CT imaging assistance. Histological images were taken with the Nanozoomer at Alafi Neuroimaging Core (S10 RR027552). VEGFAfl/fl mice from Genentech (Roche Holding AG) were kindly provided by the lab of Dr. Bjorn Olsen (Harvard). Inducible Osx Cre-ERT were generated and provided by the lab of Dr. Henry Kronenberg (Harvard). Inducible DMP1-Cre-ERT2 generated by the lab of Dr. Paola Divieti Pajevic (Boston University) were kindly provided by the lab of Dr. Alexander Robling (Indiana University Medical School).