metastasis

Tgif1 – a novel regulator of the osteoblast-breast cancer cell crosstalk in bone metastasis
Full Text
Breast cancer preferentially metastasizes to bone and over 70% of patients with advanced disease will develop bone metastases. Survival for these patients is poor and their high morbidity is often a consequence of skeletal-related events (i.e. fractures) due to the predominantly osteolytic nature of the disease. Once tumor cells proliferate in the bone, they disturb the finely tuned balance between bone formation and resorption, resulting in the so-called “vicious cycle of bone metastasis”. Factors secreted by the tumor cells stimulate osteoclasts to excessively resorb bone which in turn results in the release of growth factors from the bone matrix that further stimulate tumor growth. Thereby the vicious cycle is driven forward. Once osteolytic lesions have developed, the disease is incurable. Although osteoclast activation is the hallmark of breast-cancer induced bone disease, our knowledge about the role of osteoblasts in this process remains limited, particularly in terms of their contribution to the earliest stages of metastasis development.
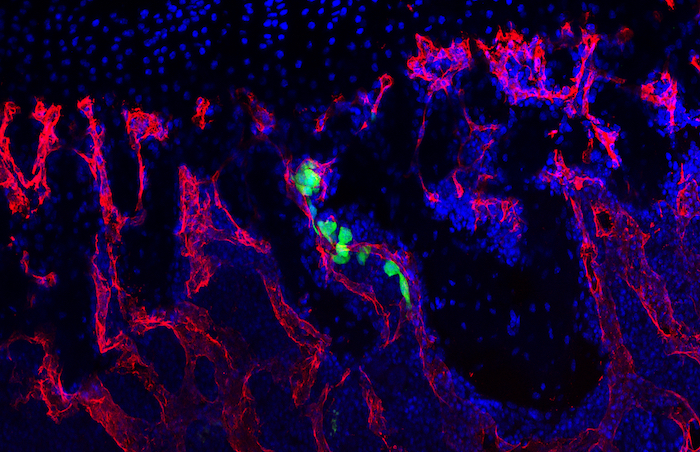
We hypothesized that osteoblasts regulate the early stages of breast cancer bone metastases, including the migration of breast cancer cells to the metastatic site. In our recently published study in Breast Cancer Research we consequently investigated the osteoblast-breast cancer cell interaction in vitro and in vivo. Indeed osteoblast-conditioned medium supported breast cancer cell migration.
We recently identified TG-interacting factor-1 (Tgif1) as a regulator of osteoblast function and bone remodeling and therefore we proposed a potential role of Tgif1 in mediating the osteoblast-breast cancer cell cross talk. Consistently we observed an increased expression of Tgif1 in osteoblasts when they were stimulated by breast cancer cells. Additionally, osteoblasts from mice lacking Tgif1 (Tgif1-/-) failed to stimulate breast cancer cell migration when compared to control (Tgif1+/+). To better understand the underlying molecular mechanisms, we performed RNA-seq analysis using osteoblasts obtained from Tgif1+/+ and Tgif1−/− mice and identified Semaphorin 3E (Sema3E) to be abundantly expressed by Tgif1-/- osteoblasts. We then confirmed that increasing concentrations of Sema3E impaired breast cancer cell migration. These findings suggest that Tgif1 in osteoblasts supports breast cancer cell migration by suppressing Sema3E expression.
Our observations then raised the question of whether Tgif1 might also be implicated in the initiation of metastatic bone disease in vivo. To explore this hypothesis, we injected 4T1 breast cancer cells into mice with lacking Tgif1 or control littermates. Indeed we observed an attenuated metastatic burden in Tgif1-deficient mice. This might at least in part be mediated by osteoblasts as Tgif1-/- mice had significantly reduced numbers of osteoblasts when compared to control mice.
In summary, our recent work identified Tgif1 as a novel regulator of the osteoblast-breast cancer cell interaction. Furthermore, we propose that Tgif1 in the bone microenvironment is implicated in the establishment and progression of breast cancer bone metastases and might therefore provide novel therapeutic opportunities to treat the initiation and progression of breast cancer metastasis to bones.
These findings are described in an article titled Breast cancer bone metastases are attenuated in a Tgif1-deficient bone microenvironment, published in the journal Breast Cancer Research. This work was performed by Marie-Therese Haider, Hiroaki Saito, Jennifer Zarrer, Kevin Uzhunnumpuram, Sankari Nagarajan, Vijayalakshmi Kari, Michael Horn-Glander, Stefan Werner, Eric Hesse and Hanna Taipaleenmäki. Modified content and image are licensed under a Creative Commons Attribution 4.0 International License.
Capitalising on the use of bone-modulating drugs in the treatment of breast cancer metastasis

Infographic

Abstract
Most breast cancer patients have no evidence of metastasis at the time of original diagnosis, yet ~30% of patients experience recurrent disease. Over 90% of cancer deaths occur due to metastasis, and bone is the most common site of breast cancer metastasis. In some clinical trials, adjuvant zoledronic acid (ZA), a bone-targeting agent, increased disease-free survival but responses were nevertheless limited. The reasons for limited responses are unknown and the mechanisms underlying ZA’s protective effect are unclear. Here, using preclinical breast cancer metastasis models, we establish that bone marrow hematopoietic cells harbor tumor suppressive activity in response to ZA. Specifically, ZA renders myeloid/osteoclast progenitor cells (M/OCPs) tumor-suppressive by altering their gene expression profile and lineage potential. Granulocyte-colony stimulating factor (G-CSF) counteracts ZA’s beneficial effects by directing M/OCP differentiation toward osteoclasts, which ablates metastasis suppression. Women enrolled in a clinical trial who had plasma G-CSF levels >23 pg/mL at randomization experienced a significant reduction in disease-free survival with adjuvant ZA. Our study lays a foundation for understanding breast cancer patient responses to ZA and suggests that finding ways to capitalize on M/OCP function and differentiation potential would constitute novel therapeutic approaches to prevent or limit metastatic disease in the bone.
Thesis
The full text of Dr Jessalyn Ubellacker’s thesis is unavailable.
Imaging of prostate cancer dormancy in the skeleton
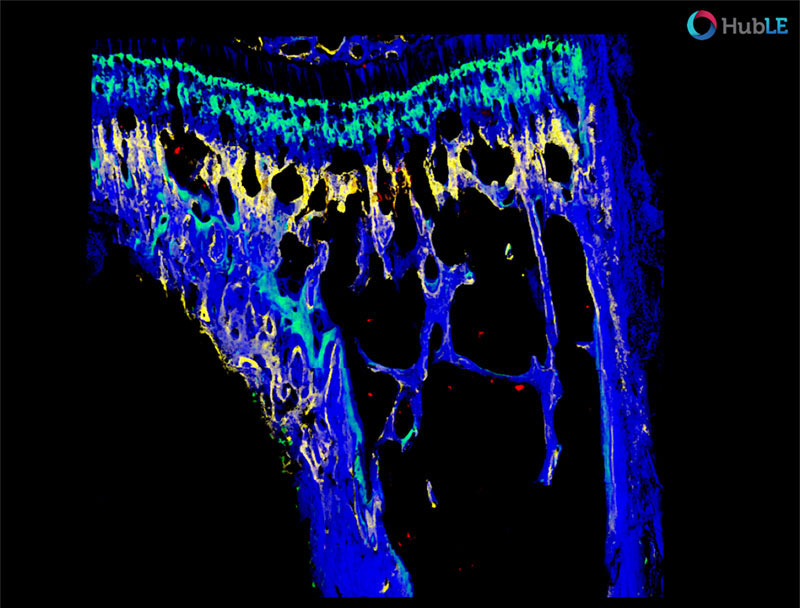
Multi-photo microscopy rendering of prostate cancer cells (red, bone (blue), alizarin complexion (yellow) and calcium (green) at mouse distal tibia.