osteoblast

VEGFA’s critical role in blood vessel formation during bone repair is cell type and injury specific
Full Text
Formation of new blood vessels is necessary for proper bone fracture healing. In support of this, fracture patients with pre-existing conditions associated with vascular disease and injury, such as smoking, advanced age, diabetes or other large-scale soft tissue trauma have lower successful fracture healing rates. Vascular endothelial growth factor A, abbreviated as VEGFA, is essential in coordinating the proliferation and recruitment of existing blood vessels across a range of bodily injury sites. In bone, blocking VEGFA with certain pharmacological agents, has been shown to inhibit bone fracture healing. However, since these pharmacological approaches are systemic and nonspecific, the cells in bone that make VEGFA and are necessary for blood vessel formation during fracture healing, remain poorly defined. This work is important to understanding why certain fractures won’t heal and would inform future treatment strategies.
Recent work in the role of blood vessels in bone came from Henry Kronenberg and Crista Maes at Harvard. These authors demonstrated that VEGFA expressing Osterix (Osx) lineage cells, a marker of early osteoprogenitors which give rise to bone forming cells called osteoblasts, secrete VEGFA and invade the fracture callus with blood vessels. Furthermore, the team of Bjorn Olsen at Harvard, demonstrated via genetic deletion of VEGFA from Osx lineage cells that blood vessel formation during cortical defect healing was impaired. However, this localized bone injury doesn’t recapitulate all aspects of fracture repair, including periosteal activation in the outer lining of the bone. Building upon these important works, our lab group at WUSTL decided to test the role of VEGFA from osteoprogenitors (Osx lineage) and their more mature cells osteoblasts and osteocytes, marked by Dentin Matrix Protein 1 (DMP1), during clinical fracture repair models such as stress fractures and full fracture.
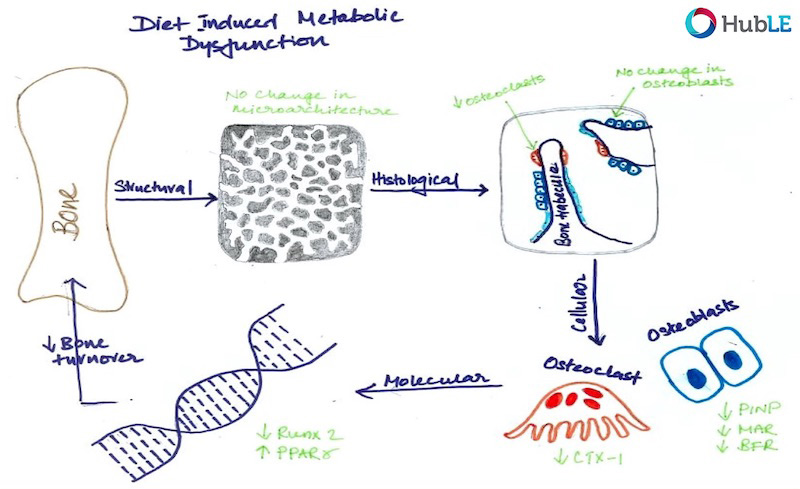
In our study, published in JBMR in 2019, we performed genetic inducible deletion of VEGFA in different cell types. We deleted VEGFA from all cells, or from bone-forming osteoblast cells at 2 stages of maturation; osteoprogenitors and mature osteoblasts/osteocytes. We then performed either a full semi-stabilized fracture or stress fracture. Cortical defect healing was also tested to compare to the results of the Olsen team. Our study revealed that loss of VEGFA in all cells blocked bone and blood vessel formation regardless of the injury model, reaffirming previous pharmacological results. More importantly, we showed that VEGFA from osteoprogenitor cells (Osx lineage) but not more mature osteoblasts/osteocytes (DMP1 lineage) was necessary for maximal bone formation and blood vessel formation in stress fracture and full fracture models. Surprisingly, VEGFA from osteoprogenitor cells at the time of injury was not important for cortical defect repair. These results together, highlight that the VEGFA cell source necessary for bone healing depends on the type of bone injury. Furthermore, our study is the first to indicate that VEGFA from different osteoblast maturation stages play nonredundant roles during clinical models of fracture healing. Our results may also help explain why recombinant BMPs, which stimulate VEGFA production in osteoprogenitors, are promising treatments to induce bone formation during fracture. Overall, these findings highlight that osteoprogenitors are critically sources of VEGFA during fracture healing. Furthermore, these same cells should be clinical targets for improving bone blood vessel formation to promote fracture repair in patients with poor healing outcomes.
These findings are described in more detail in the article entitled “VEGFA From Early Osteoblast Lineage Cells (Osterix+) Is Required in Mice for Fracture Healing” published in JBMR. The work was conducted by Evan Buettmann, Jennifer McKenzie, Nicole Migotsky, David Sykes, Pei Hu, Susumu Yoneda, Matthew Silva. This work was funded by NIAMS (R01 AR050211 and P30 AR057235). The authors would like to thank the Washington University in St. Louis Musculoskeletal Research Center (MRC) Cores and staff for histological and micro-CT imaging assistance. Histological images were taken with the Nanozoomer at Alafi Neuroimaging Core (S10 RR027552). VEGFAfl/fl mice from Genentech (Roche Holding AG) were kindly provided by the lab of Dr. Bjorn Olsen (Harvard). Inducible Osx Cre-ERT were generated and provided by the lab of Dr. Henry Kronenberg (Harvard). Inducible DMP1-Cre-ERT2 generated by the lab of Dr. Paola Divieti Pajevic (Boston University) were kindly provided by the lab of Dr. Alexander Robling (Indiana University Medical School).
Tgif1 – a novel regulator of the osteoblast-breast cancer cell crosstalk in bone metastasis
Full Text
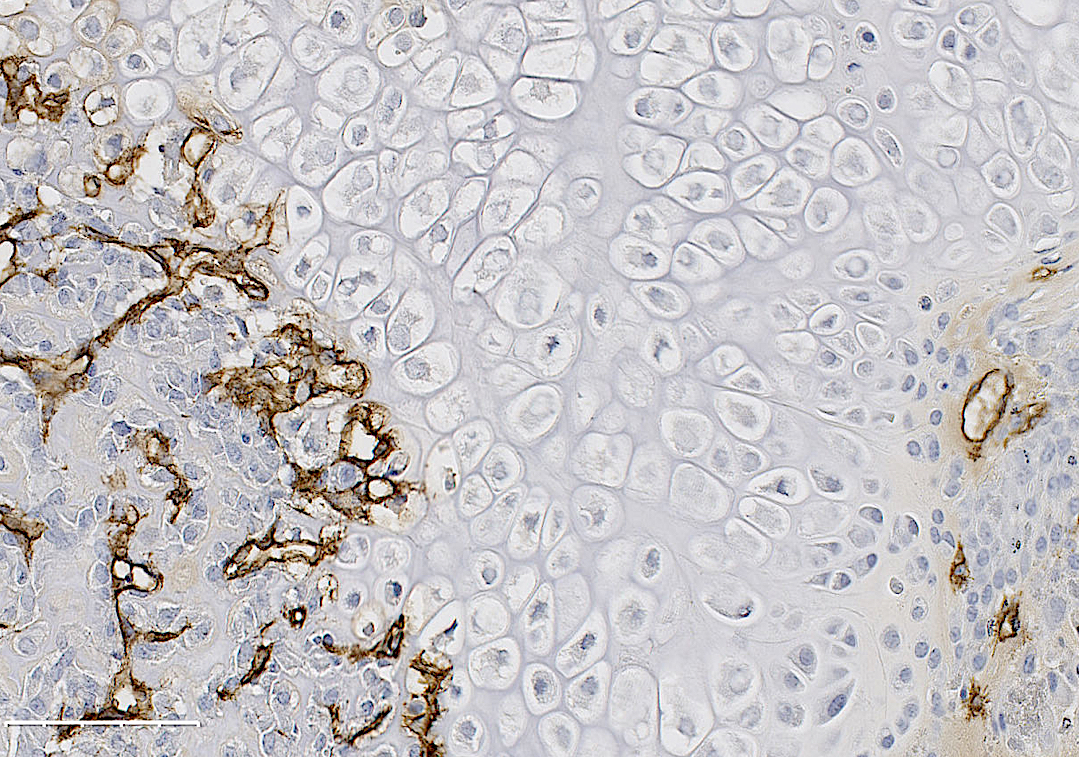
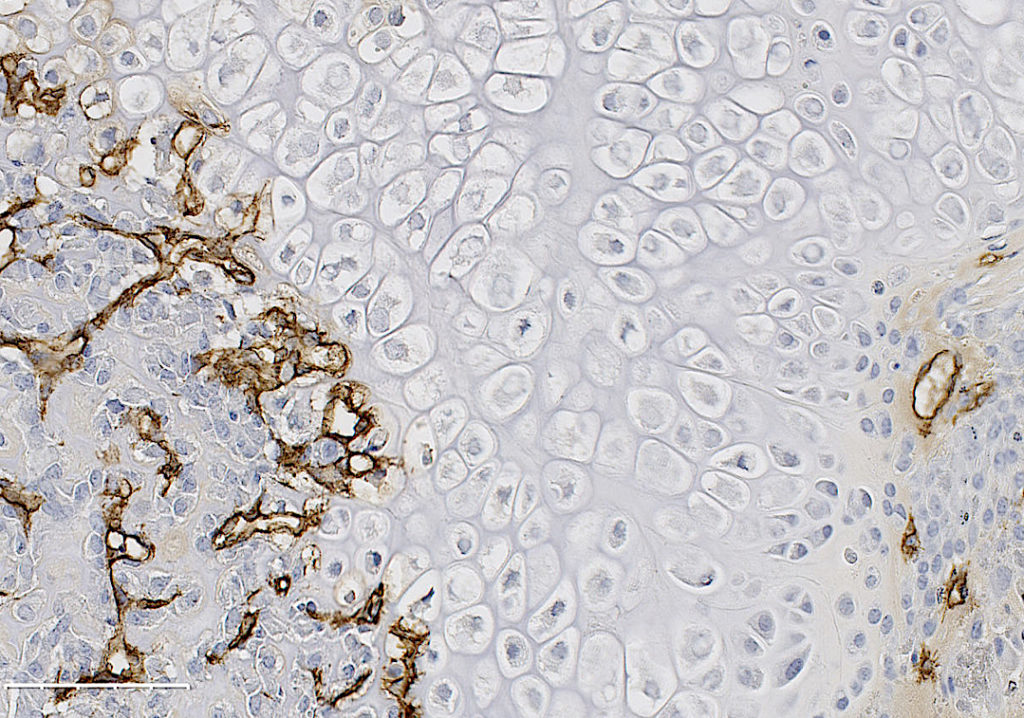
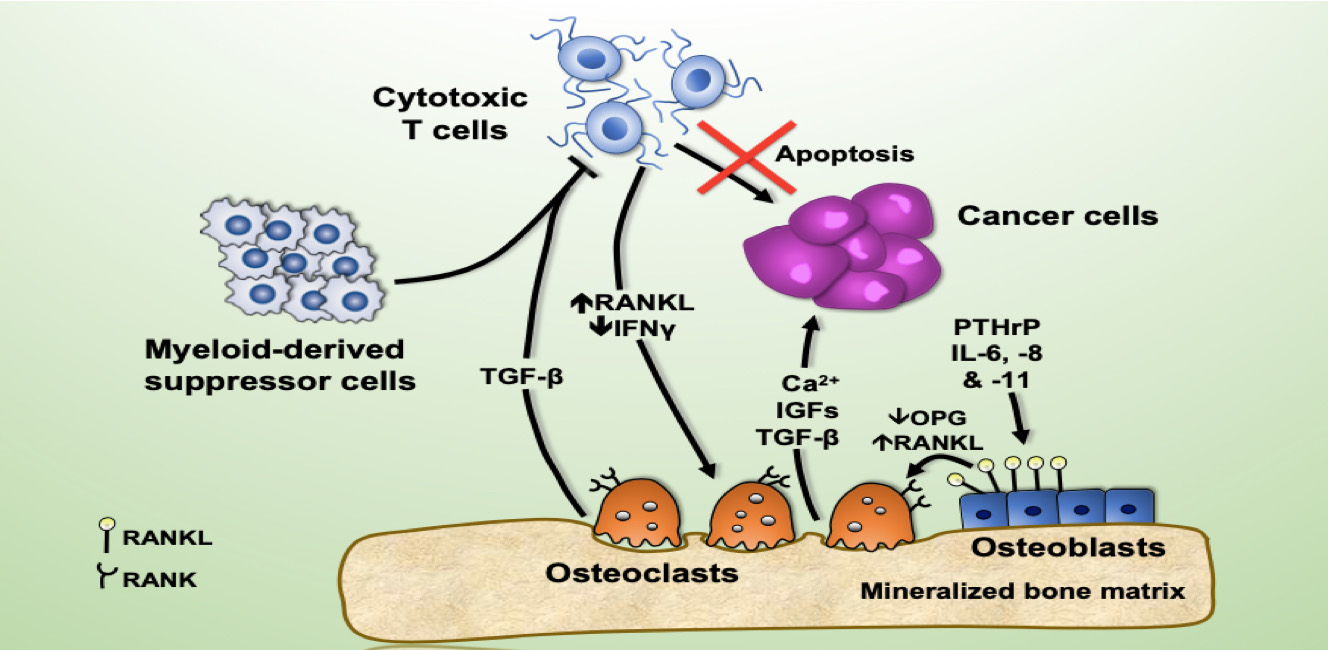
Breast cancer preferentially metastasizes to bone and over 70% of patients with advanced disease will develop bone metastases. Survival for these patients is poor and their high morbidity is often a consequence of skeletal-related events (i.e. fractures) due to the predominantly osteolytic nature of the disease. Once tumor cells proliferate in the bone, they disturb the finely tuned balance between bone formation and resorption, resulting in the so-called “vicious cycle of bone metastasis”. Factors secreted by the tumor cells stimulate osteoclasts to excessively resorb bone which in turn results in the release of growth factors from the bone matrix that further stimulate tumor growth. Thereby the vicious cycle is driven forward. Once osteolytic lesions have developed, the disease is incurable. Although osteoclast activation is the hallmark of breast-cancer induced bone disease, our knowledge about the role of osteoblasts in this process remains limited, particularly in terms of their contribution to the earliest stages of metastasis development.
We hypothesized that osteoblasts regulate the early stages of breast cancer bone metastases, including the migration of breast cancer cells to the metastatic site. In our recently published study in Breast Cancer Research we consequently investigated the osteoblast-breast cancer cell interaction in vitro and in vivo. Indeed osteoblast-conditioned medium supported breast cancer cell migration.
We recently identified TG-interacting factor-1 (Tgif1) as a regulator of osteoblast function and bone remodeling and therefore we proposed a potential role of Tgif1 in mediating the osteoblast-breast cancer cell cross talk. Consistently we observed an increased expression of Tgif1 in osteoblasts when they were stimulated by breast cancer cells. Additionally, osteoblasts from mice lacking Tgif1 (Tgif1-/-) failed to stimulate breast cancer cell migration when compared to control (Tgif1+/+). To better understand the underlying molecular mechanisms, we performed RNA-seq analysis using osteoblasts obtained from Tgif1+/+ and Tgif1−/− mice and identified Semaphorin 3E (Sema3E) to be abundantly expressed by Tgif1-/- osteoblasts. We then confirmed that increasing concentrations of Sema3E impaired breast cancer cell migration. These findings suggest that Tgif1 in osteoblasts supports breast cancer cell migration by suppressing Sema3E expression.
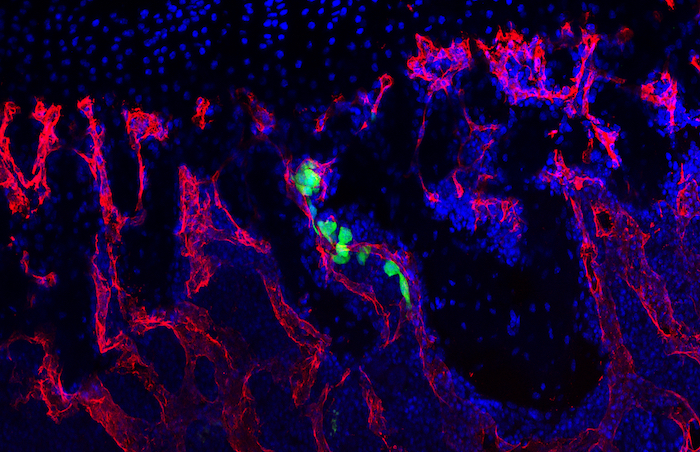
Our observations then raised the question of whether Tgif1 might also be implicated in the initiation of metastatic bone disease in vivo. To explore this hypothesis, we injected 4T1 breast cancer cells into mice with lacking Tgif1 or control littermates. Indeed we observed an attenuated metastatic burden in Tgif1-deficient mice. This might at least in part be mediated by osteoblasts as Tgif1-/- mice had significantly reduced numbers of osteoblasts when compared to control mice.
In summary, our recent work identified Tgif1 as a novel regulator of the osteoblast-breast cancer cell interaction. Furthermore, we propose that Tgif1 in the bone microenvironment is implicated in the establishment and progression of breast cancer bone metastases and might therefore provide novel therapeutic opportunities to treat the initiation and progression of breast cancer metastasis to bones.
These findings are described in an article titled Breast cancer bone metastases are attenuated in a Tgif1-deficient bone microenvironment, published in the journal Breast Cancer Research. This work was performed by Marie-Therese Haider, Hiroaki Saito, Jennifer Zarrer, Kevin Uzhunnumpuram, Sankari Nagarajan, Vijayalakshmi Kari, Michael Horn-Glander, Stefan Werner, Eric Hesse and Hanna Taipaleenmäki. Modified content and image are licensed under a Creative Commons Attribution 4.0 International License.
Mechanobiological changes during estrogen deficiency leading to bone loss
Infographic
Abstract
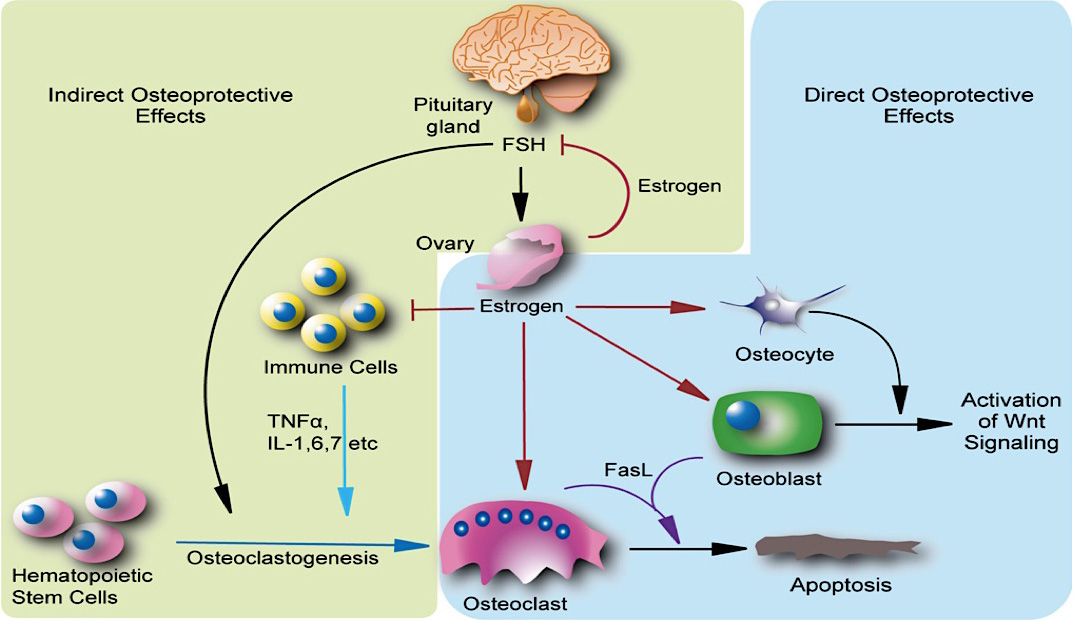
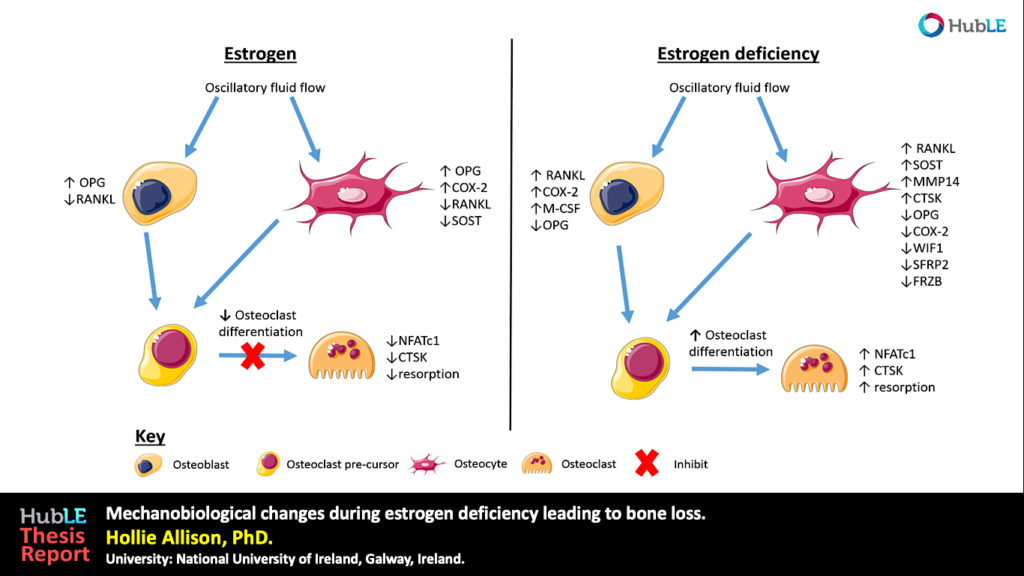
Osteoporosis, which predominantly occurs in females, affects 200 million women worldwide and is characterised by bone loss and microarchitectural degradation, which leads to fractures, pain and immobility. Conventional treatments for osteoporosis target bone loss by inhibiting osteoclast activity, but these only prevent osteoporotic fractures in 50% of sufferers. An advanced understanding of the underlying mechanisms and treatments for osteoporosis is required to avert the projected trend, whereby the worldwide economic burden of treatment will reach $132 billion by 2050. Osteoblasts and osteocytes are known to regulate the differentiation, function and survival of osteoclasts, through expression of paracrine factors, which can be altered following mechanical loading and are affected by the presence of estrogen. Studies have recently revealed changes in osteoblast and osteocyte mechanobiology during estrogen deficiency, but, how these changes effect osteoclastogenesis is not fully understood. The first study of this thesis sought to assess changes in osteoblast-induced osteoclastogenesis under combined estrogen deficiency and mechanical loading, using a combination of conditioned media and co-culture experiments. MC3T3-E1 cells were cultured with pre-menopausal levels of estrogen which was then withdrawn from the media and the osteoblast like-cells were exposed to oscillatory fluid flow. These studies found that conditioned media from estrogen-treated MC3T3-E1 cells inhibited the differentiation of RAW264.7 cells and inhibited podosome belt formation. Estrogen deficient osteoblasts down-regulated osteoprotegerin (OPG) expression in response to fluid flow when compared to estrogen-treated osteoblasts, leading to a significant increase in the ratio of RANKL/OPG gene expression. Subsequently, an increase in osteoclast number and an up-regulation in nuclear factor of activated T cells cytoplasmic 1 (NFATc1) and Cathepsin K expression was observed in RAW264.7 cells. In addition, estrogen deficient osteoblasts co-cultured with RAW264.7 cells resulted in an increase in osteoclastogenesis and matrix degradation compared to co-cultures with estrogen-treated osteoblasts. Additionally, it was found that Rho-ROCK inhibition in osteoblasts, exacerbated the increase in osteoblast-induced osteoclastogenesis that arose under estrogen deficiency. It is proposed that the reduction in OPG production by mechanically stimulated osteoblasts during estrogen deficiency may leave osteoclast differentiation and matrix degradation activity unchecked and, thereby, play an important role in bone loss during osteoporosis. The second study sought to establish whether mechanically stimulated osteocytes induce osteoclastogenesis and bone resorption during estrogen deficiency. This study built upon the post-menopausal model and co-culture methods established in the first study. The findings revealed that under postmenopausal conditions RANKL/OPG gene expression is increased in mechanically stimulated osteocytes (OCY454 cells) compared to estrogen-treated osteocytes and a significant increase in osteocyte-induced osteoclast formation (by bone marrow macrophage cells) occurs leading to increased resorption. Estrogen deficient osteocytes also up-regulated sclerostin expression following mechanical loading. Interestingly, Wnt antagonists WIF1 and FRZB were down-regulated in estrogen deficient osteocytes following loading. A second aim of this study was to investigate whether a neutralising antibody against sclerostin (Scl-Ab) could revert osteocyte-mediated osteoclastogenesis and resorption by attenuating RANKL/OPG gene expression. It was demonstrated that administration of the Scl-Ab reduced pro-osteoclastogenic signalling (RANKL/OPG) between osteocytes and osteoclasts, which led to reduced resorption. Osteocytes are housed within lacunae and their dendrites extend through canaliculae to enable the formation of gap junctions with neighbouring osteocytes and osteoblasts. The lacunar-canalicular network and vascular pores facilitate movement of interstitial fluid, which confers mechanical stimulation on osteocytes and facilitates transport of nutrients and signalling molecules to and from the osteocyte. There is some evidence that this microporous network is altered during estrogen deficiency, which might alter fluid flow through the network and the ensuing responses of osteoblasts and osteocytes. While previous studies have investigated early-stage estrogen deficiency, the long term effects of estrogen deficiency and the time-sequence of changes in cortical bone microporosity are not known. For this reason, the final study of this thesis sought to determine the temporal changes that occur in the cortical vasculature and the lacunar-canalicular network during short and longer term estrogen deficiency. This study quantified microporosity and lacunar occupancy in an ovariectomised rat model of osteoporosis by micro-CT analysis, backscatter electron imaging and histological analysis. It was shown that initial increases in canalicular diameter and vascular porosity arose during short term estrogen deficiency (week 4 OVX) compared to aged-matched controls, but that these were reduced along with a decrease in lacunar diameter and lacunar occupancy in long term estrogen deficiency (week 14 OVX). These changes could be explained by perilacunar remodelling, micropetrosis or a mechanobiological adaptive response, such as increases in bone mass and bone mineralisation that may occur to alter the mechanical environment in an attempt to restore tissue homeostasis. This study also demonstrated that administration of Scl-Ab to ovariectomised rats resulted in a significant decrease in osteoclast number and prevented the occurrence of empty lacunae in long term estrogen deficiency (week 14 OVX) compared to untreated week 14 OVX animals. Together, the studies in this thesis found that the inhibitory effects mechanically loading has on pro-osteoclastogenic signalling in osteocytes and osteoblasts is attenuated during estrogen deficiency. Using cell culture and mechanobiological techniques, the changes in paracrine factor expression in estrogen deficient osteoblasts and osteocytes and their implications in terms of osteoclastogenesis and resorption were elucidated. Additionally, the temporal changes in cortical microporosity that occur were established. Collectively, the research presented in this thesis provides an important, but previously unrecognised, insight into the mechanobiological changes that occur during estrogen deficiency leading to bone loss. The information gained from this work may inform future mechanobiological treatments for osteoporosis.
Thesis
Full text of D. Allison’s thesis is available here.
Culture of the IDG-SW3 osteocyte cell line
Full Text
Introduction
The IDG-SW3 osteoblast-to-late-osteocyte cell line is derived from a temperature-sensitive DMP1-GFP transgenic mouse. IDG-SW3s cultured with interferon g (IFNγ) at 33oC will proliferate, whilst culture in mineralising conditions without IFNγ at 37oC enhances differentiation. This HubLE Method describes the protocol for culturing this cell line [1].
Materials
- Rat-tail type 1 collagen
- Recombinant mouse interferon g (IFNγ)
- Phosphate buffered saline (PBS)
- Proliferation medium Alpha Modified Essential Medium (ProlifαMEM) [Tip No. 1]
- Osteocyte-differentiation αMEM (OcyMEM) [Tip No. 2]
- Trypsin-EDTA (0.25%)
- Glutaraldehyde (2.5%)
- Alizarin Red (1%)
Methods [Update]
All procedures should be performed under sterile conditions.
- Coat all required tissue culture plastics for 1 hour at room temperature with 0.15mg/ml of rat-tail type 1 collagen in 0.02M acetic acid.
- Remove the collagen solution and either wash with PBS for immediate use, or air dry plates prior to storage [Tip No. 3].
- Thaw a vial of IDG-SW3 cells into 5ml ProlifαMEM and spin at 1,500 rpm for 5 minutes.
- Remove the supernatant, re-suspend the pellet in ProlifαMEM and seed into a collagen-coated 75cm2 flask containing ProlifαMEM. Incubate at 33oC with 5% CO2.
- Once ≥80% confluent (2-3 days post-seeding), remove medium, wash with PBS and incubate with 0.25% Trypsin for 5-10 minutes to detach cells.
- Spin at 1,500g for 5 minutes and re-suspend in ProlifαMEM (1ml per flask).
- Seed cells in collagen-coated tissue culture trays or flasks (for further expansion and use of cells at next passage) in ProlifαMEM [Tip No. 4].
- Incubate at 33oC with 5% CO2 until confluent.
- At this stage, remove the ProlifαMEM medium, carefully wash the cell monolayers with PBS and add OcyMEM. Incubate at 37oC with 5% CO2.
- Culture for up to 30-35 days with half medium changes of OcyMEM every 2-3 days. Mineralisation is usually evident from ~10-14 days.
- SFix with 2.5% glutaraldehyde for 5 minutes before staining with 1% alizarin red to visualise mineralised nodules [Tip No. 5].
Tips [Update]
ProlifαMEM: Add 10% heat-inactivated foetal calf serum (FCS), AB/AM (100U/ml penicillin, 100mg/ml streptomycin, 0.25mg/ml amphotericin) and L-glutamine (200mM). Aliquot the stock media and add IFNγ (2500U/ µl). Incubate the medium at 33oC prior to use and limit exposure to heat due to IFNγ degradation.
OcyMEM: Add 10% heat-inactivated foetal calf serum (FCS), AB/AM (100U/ml penicillin, 100mg/ml streptomycin, 0.25mg/ml amphotericin) and L-glutamine (200mM). Add 50µg/ml ascorbate and 2-4mM β-glycerophosphate (the original paper uses 4mM β-GP [1], however, IDG-SW3 cells differentiate and mineralise sufficiently in 2mM). Always make fresh on the day of use.
Collagen-coated tissue culture plates/ flasks: Ensure all plates are coated under sterile conditions in a tissue culture hood. Use a cold pipette (stored in the freezer until use) to stop the collagen sticking to the plastic. The 0.15mg/ml collagen solution can be re-used 5-6 times; coating for ~1 hour each time. Coated plastics wrapped in parafilm can be stored at 4oC for up to 6 months until use.
Seeding density: IDG-SW3 cells will mineralise in 12 and 6-well plates but due to the long culture duration some monolayer peeling should be expected. Woo et al. [1] recommend seeding IDG-SW3 cells at 4×104 cells/cm2, although the lower densities of 104 (12-well) and 105 (6-well) will also support osteocyte proliferation, mineralisation and differentiation. To expand IDG-SW3 cells for the subsequent passage seed at 5×105 cells/ 75cm2 flask.
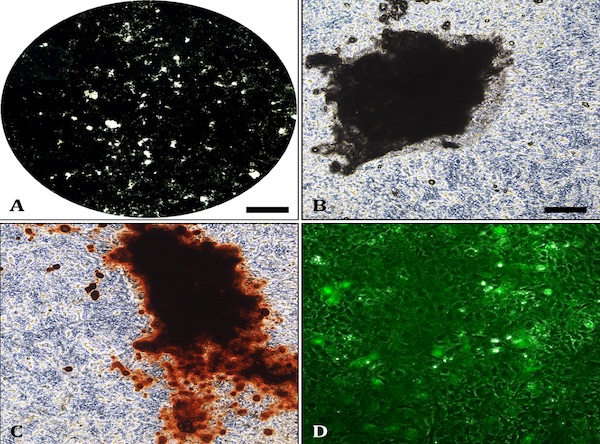
Alizarin Red staining: Mineralised bone nodules can be stained with alizarin red (Fig.1C). It is also possible to obtain good quality images on unstained cell layers (Fig 1A-1B). DMP1-GFP expression can be visually monitored throughout the differentiation process (Fig.1D). Evaluation of E11, DMP1 and sclerostin gene/ protein expression is also advisable to confirm osteocyte differentiation.
References [Update]
Woo SM, Rosser J, Dusevich V, Kalajzic I, Bonewald LF (2011). Cell line IDG-SW3 replicates osteoblast-to-late osteocyte differentiation in vitro and accelerates bone formation in vivo. J Bone Mineral Res 26:2634-2646.