BACKGROUND:
Autosomal Dominant Osteopetrosis type 2 (ADO2) is a rare genetic bone disease caused by a reduction of osteoclast function. Osteoclasts are multinucleated cells responsible for the removal of bone matrix during the process of bone resorption. In ADO2 patients, the function of these cells is impaired, inducing an increased bone mass, especially at the skull base, pelvis and vertebrae. Nevertheless, ADO2 bones are very fragile, and patients experience multiple low energy fractures during their lifetime. Other symptoms associated with ADO2 are osteomyelitis, haematological alterations and in rare cases deafness and blindness due to nerve compression.
Genetically, only mutations in the CLCN7gene have been associated to ADO2, with a penetrance of 66%. The CLCN7gene encodes for the proton/chloride antiporter ClC7 that localises mainly in the endo/lysosomal compartment and in the ruffled boarder of the osteoclasts. Its main function is to balance the electrical charges and allow a correct acidification of the intracellular vesicles and of the resorption lacuna, ensuring the maintenance of a correct electrical shunt.
To date, there is not a resolutive therapy for the disease and ADO2 patients experience a reduced quality of life.
Finally, the gene implicated in the pathogenesis of ADO2, the CLCN7gene, is expressed in many tissues, suggesting that mutations on his sheet could have a systemic impact.
AIM OF THE STUDY:
An essential tool for studying the disease and developing a potential therapy is represented by the ADO2 animal model developed in our laboratory in 2013. This mouse model, carrying the Clcn7G213Rmutation, homologue of the human CLCN7G215Rmutation, fully recapitulates the bone phenotype observed in the ADO2 patients.
Starting from the above-mentioned background and taking advantage of this animal model, my PhD project had 3 main aims:
- To progress towards the preclinical development of an siRNA-based ADO2 therapy (Chapter 2).
- To analyse the ADO2 mice phenotype beyond the bone (Chapter 3).
- To study the effect of the G213R mutation on the subcellular localization of ClC7 (Chapter 3).
RESULTS:
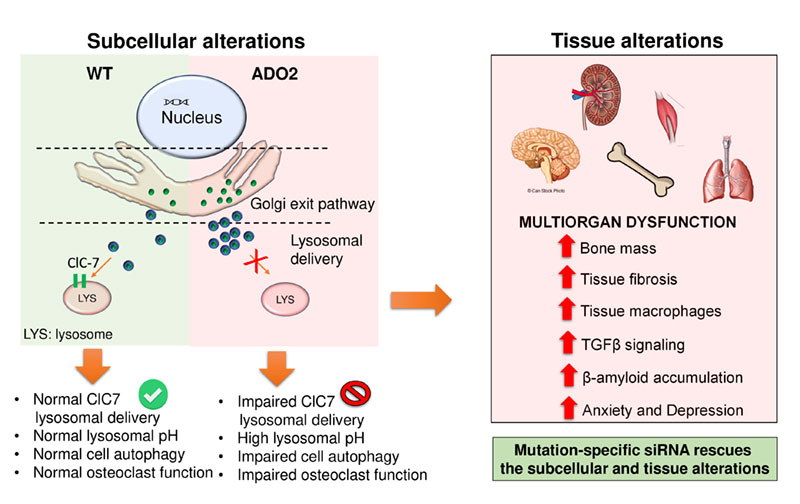
Our work demonstrated that ADO2 cannot be considered a “pure” bone disease, but it has a systemic effect. Indeed, we found that the Clcn7was expressed in several organs, except for the muscle. Based on the Clcn7expression, we performed an RNA deep sequencing (RNA dSeq) analysis in lungs, kidneys and muscles, as examples of tissues that express the gene at high, medium and low level, respectively. Data obtained from the RNA dSeq demonstrated an enrichment of the genes involved in both the onset of tissue fibrosis and the biology of macrophages in lungs, kidneys and muscles of ADO2 mice compare to their WT littermates. Histopathological analyses further confirmed the presence of perivascular fibrosis along with an increased number of tissue macrophages in the ADO2 organs. Moreover, bioinformatic analyses revealed the presence of 10 altered signalling pathways shared by the ADO2 lungs, kidneys and muscles. Among them, we focused our attention on the TGFβ pathway, known to be involved in tissue fibrosis and macrophages` biology. Real time RT-PCR, western blot and immunofluorescence confirmed the activation of TGFβ signalling in lungs, kidneys and muscles, suggesting a potential role for this pathway in the multiorgan dysfunction.
In addition, we observed the presence of behavioural alterations in our ADO2 mice. We found an increased anxiety and depression in ADO2 mice compare to their WT littermates, along with an increased accumulation of β-amyloids and an alteration in the distribution of glial cells. The RNA dSeq confirmed the presence of an enrichment in pathways involved in long-term depression, long-term potentiation and neuroactive ligand-receptor interaction.
Our results were further strengthened by the observation that a small cohort of ADO2 patients presents with extra skeletal alterations.
At subcellular level, we found that the Clcn7G213Rmutation was able to alter the localisation of the ClC7 protein. Indeed, we found that the protein was trapped mainly in the Golgi and in the clathrin coated vesicles of the ADO2 bone marrow mononuclear cells (BMMCs). Consequently, its localisation in the lysosomes was reduced in ADO2 BMMCs along with an altered intracellular acidity and cell autophagy. The same results were found in ADO2 osteoclasts and in the tissues affected by the disease, including lungs, kidneys, muscles and brain.
Finally, another aim of my PhD project was to progress toward the preclinical development of a siRNA-based ADO2 therapy. Our results showed that the use of a mutation-specific siRNA was able to revert the bone phenotype in the ADO2 mice. We tested intraperitoneal and subcutaneous routes of administration, at different ages and in both genders. All the regimes tested were effective. Moreover, we demonstrated that our siRNA-based approach was also able to treat the extra-skeletal alterations in ADO2 mice. Overall, we demonstrated that our patented siRNA-based therapy for the ADO2 (patent application PCT/IB2015/053730) was able to systemically treat the ADO2 mice, thus fully curing the disease.
CONCLUSION:
The work done during my PhD allows us to understand that ADO2 is a systemic disease affecting several organs and to develop a target therapy able to treat both the bone and the non-bone alterations seen in ADO2 mice.
