Infographic
Abstract
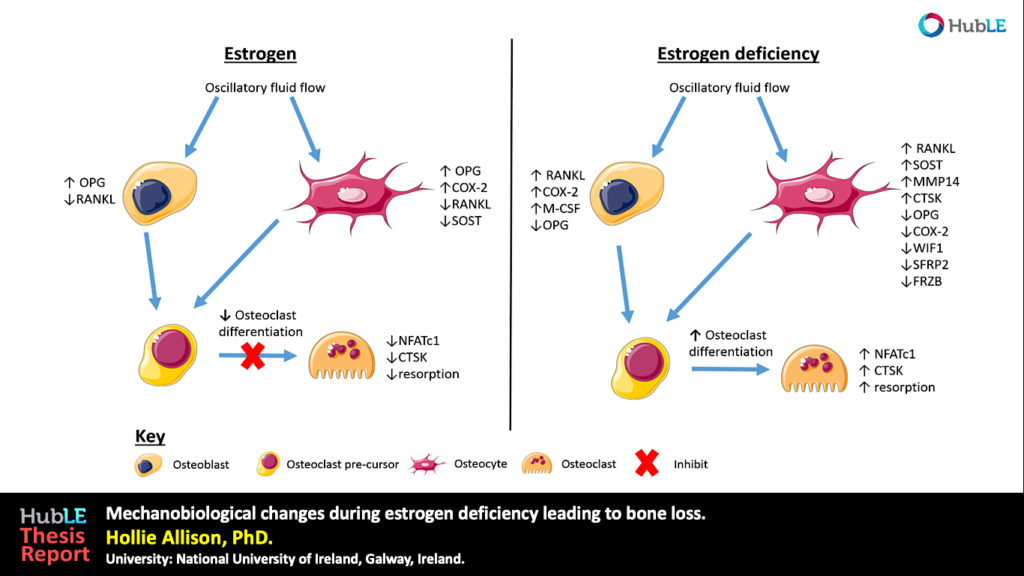
Osteoporosis, which predominantly occurs in females, affects 200 million women worldwide and is characterised by bone loss and microarchitectural degradation, which leads to fractures, pain and immobility. Conventional treatments for osteoporosis target bone loss by inhibiting osteoclast activity, but these only prevent osteoporotic fractures in 50% of sufferers. An advanced understanding of the underlying mechanisms and treatments for osteoporosis is required to avert the projected trend, whereby the worldwide economic burden of treatment will reach $132 billion by 2050. Osteoblasts and osteocytes are known to regulate the differentiation, function and survival of osteoclasts, through expression of paracrine factors, which can be altered following mechanical loading and are affected by the presence of estrogen. Studies have recently revealed changes in osteoblast and osteocyte mechanobiology during estrogen deficiency, but, how these changes effect osteoclastogenesis is not fully understood. The first study of this thesis sought to assess changes in osteoblast-induced osteoclastogenesis under combined estrogen deficiency and mechanical loading, using a combination of conditioned media and co-culture experiments. MC3T3-E1 cells were cultured with pre-menopausal levels of estrogen which was then withdrawn from the media and the osteoblast like-cells were exposed to oscillatory fluid flow. These studies found that conditioned media from estrogen-treated MC3T3-E1 cells inhibited the differentiation of RAW264.7 cells and inhibited podosome belt formation. Estrogen deficient osteoblasts down-regulated osteoprotegerin (OPG) expression in response to fluid flow when compared to estrogen-treated osteoblasts, leading to a significant increase in the ratio of RANKL/OPG gene expression. Subsequently, an increase in osteoclast number and an up-regulation in nuclear factor of activated T cells cytoplasmic 1 (NFATc1) and Cathepsin K expression was observed in RAW264.7 cells. In addition, estrogen deficient osteoblasts co-cultured with RAW264.7 cells resulted in an increase in osteoclastogenesis and matrix degradation compared to co-cultures with estrogen-treated osteoblasts. Additionally, it was found that Rho-ROCK inhibition in osteoblasts, exacerbated the increase in osteoblast-induced osteoclastogenesis that arose under estrogen deficiency. It is proposed that the reduction in OPG production by mechanically stimulated osteoblasts during estrogen deficiency may leave osteoclast differentiation and matrix degradation activity unchecked and, thereby, play an important role in bone loss during osteoporosis. The second study sought to establish whether mechanically stimulated osteocytes induce osteoclastogenesis and bone resorption during estrogen deficiency. This study built upon the post-menopausal model and co-culture methods established in the first study. The findings revealed that under postmenopausal conditions RANKL/OPG gene expression is increased in mechanically stimulated osteocytes (OCY454 cells) compared to estrogen-treated osteocytes and a significant increase in osteocyte-induced osteoclast formation (by bone marrow macrophage cells) occurs leading to increased resorption. Estrogen deficient osteocytes also up-regulated sclerostin expression following mechanical loading. Interestingly, Wnt antagonists WIF1 and FRZB were down-regulated in estrogen deficient osteocytes following loading. A second aim of this study was to investigate whether a neutralising antibody against sclerostin (Scl-Ab) could revert osteocyte-mediated osteoclastogenesis and resorption by attenuating RANKL/OPG gene expression. It was demonstrated that administration of the Scl-Ab reduced pro-osteoclastogenic signalling (RANKL/OPG) between osteocytes and osteoclasts, which led to reduced resorption. Osteocytes are housed within lacunae and their dendrites extend through canaliculae to enable the formation of gap junctions with neighbouring osteocytes and osteoblasts. The lacunar-canalicular network and vascular pores facilitate movement of interstitial fluid, which confers mechanical stimulation on osteocytes and facilitates transport of nutrients and signalling molecules to and from the osteocyte. There is some evidence that this microporous network is altered during estrogen deficiency, which might alter fluid flow through the network and the ensuing responses of osteoblasts and osteocytes. While previous studies have investigated early-stage estrogen deficiency, the long term effects of estrogen deficiency and the time-sequence of changes in cortical bone microporosity are not known. For this reason, the final study of this thesis sought to determine the temporal changes that occur in the cortical vasculature and the lacunar-canalicular network during short and longer term estrogen deficiency. This study quantified microporosity and lacunar occupancy in an ovariectomised rat model of osteoporosis by micro-CT analysis, backscatter electron imaging and histological analysis. It was shown that initial increases in canalicular diameter and vascular porosity arose during short term estrogen deficiency (week 4 OVX) compared to aged-matched controls, but that these were reduced along with a decrease in lacunar diameter and lacunar occupancy in long term estrogen deficiency (week 14 OVX). These changes could be explained by perilacunar remodelling, micropetrosis or a mechanobiological adaptive response, such as increases in bone mass and bone mineralisation that may occur to alter the mechanical environment in an attempt to restore tissue homeostasis. This study also demonstrated that administration of Scl-Ab to ovariectomised rats resulted in a significant decrease in osteoclast number and prevented the occurrence of empty lacunae in long term estrogen deficiency (week 14 OVX) compared to untreated week 14 OVX animals. Together, the studies in this thesis found that the inhibitory effects mechanically loading has on pro-osteoclastogenic signalling in osteocytes and osteoblasts is attenuated during estrogen deficiency. Using cell culture and mechanobiological techniques, the changes in paracrine factor expression in estrogen deficient osteoblasts and osteocytes and their implications in terms of osteoclastogenesis and resorption were elucidated. Additionally, the temporal changes in cortical microporosity that occur were established. Collectively, the research presented in this thesis provides an important, but previously unrecognised, insight into the mechanobiological changes that occur during estrogen deficiency leading to bone loss. The information gained from this work may inform future mechanobiological treatments for osteoporosis.
Thesis
Full text of D. Allison’s thesis is available here.