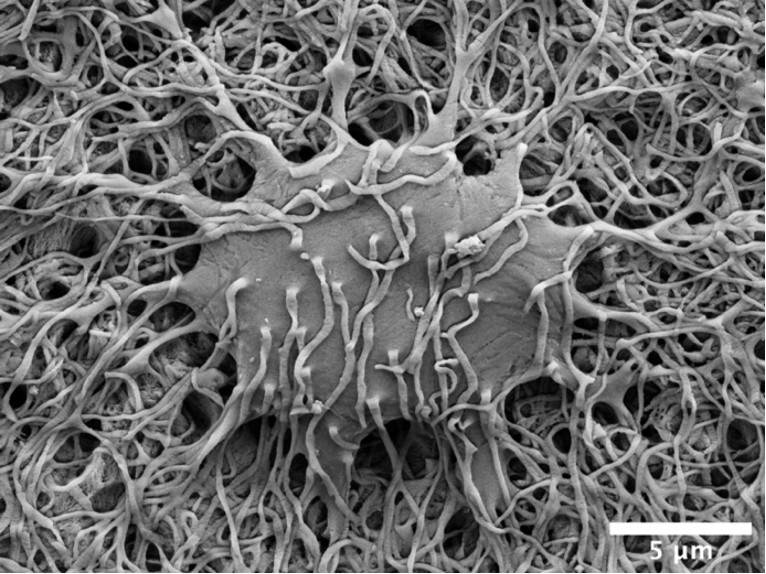
The osteocyte lacuno-canalicular network is a fundamental structural and functional component of bone. The osteocytes play the vital role of mechanosensory cells by orchestrating bone remodeling. Furthermore, the morphology of the osteocyte lacuno-canalicular network (e.g., osteocyte interconnectivity) reflects the underlying mechanobiological status of bone, which is relevant for understanding the processes of bone growth and repair/renewal. To better characterize this complex morphology, novel high-resolution imaging techniques have been implemented, including tissue specimens from various types of implanted biomaterials. Of the various imaging techniques available, resin cast etching is a straightforward method involving sequential acid etching and alkali digestion of resin embedded bone to visualize the osteocyte lacuno-canalicular network using scanning electron microscopy.
This technique was first proposed in 1985, where Todd Curtis and co-workers used resin cast etching to first characterize trabecular and cortical bone morphology we all have come to know today. They did this by visualizing the osteocytes as flattened oval discs aligned parallel to the successive layers of collagen. Today, this technique has been applied across the spectrum of research, from developmental biology to the impact of genetic alterations on bone, ageing, spinal cord injury and long-term immobilization, to the effects of cancer on bone, tissue engineering, and finally, to drug discovery. Two unique applications of resin cast etching in the clinical and experimental biomaterials research field include: 1) visualizing the distribution of osteocytes around metal implants used in dentistry, craniomaxillofacial surgery, and orthopedics; and 2) determining the direction of bone formation around resorbable biomaterials designed to regenerate bone lost due to trauma or surgery (e.g., calcium phosphates). Resin cast etching has revealed the three-dimensional morphology of osteocytes and their lacuno-canalicular network, providing evidence of alterations in network interconnectivity due to changes in either osteocyte density—the number of osteocytes per unit area—or dendricity—the number of canaliculi per osteocyte lacuna. Furthermore, resin cast etching is a convenient way to characterize lacunar shape and size, which are important markers of development, aging, and bone regeneration. Ultimately, the quality of information gathered from the resin cast is dependent on sample preparation. In some situations (e.g., a heterogeneously dense specimen), sub-optimal infiltration and polymerization of the embedding medium results in areas of bone that falsely appear devoid of the osteocyte lacuno-canalicular network, and alternative imaging techniques will need to be considered.
Resin cast etching offers advantages over other techniques that are commonly used to examine the osteocyte lacuno-canalicular system. Most notably, any resin embedded specimen, such as clinical biopsies, can be used for resin cast etching without the need for additional contrast staining. The added convenience of performing additional assessments of bone mineral density distribution and elemental analysis—all within the same scanning electron microscope—make resin cast etching ideal for gathering both quantitative and qualitative information of the osteocyte morphology. One such approach in the bone repair biomaterials community has used resin cast etching to examine the bone response in novel biomaterials applications, including those biomaterials that gradually degrade in vivo and/or possess the capability to deliver substances that impact bone metabolism (e.g., bone morphogenetic proteins). Over time, these types of investigations are expected to stimulate the development and validation of advanced therapeutic strategies targeting pathological and rare bone diseases in addition to commonly encountered conditions such as osteoporosis.
The authors wish to thank the Japan Society for the Promotion of Science (JSPS; grant number 17K17564), Takeda Science Foundation, the Svenska Sällskapet för Medicinsk Forskning (SSMF), the Hjalmar Svensson Foundation, and the Kungliga Vetenskaps-och Vitterhets-Samhället i Göteborg.