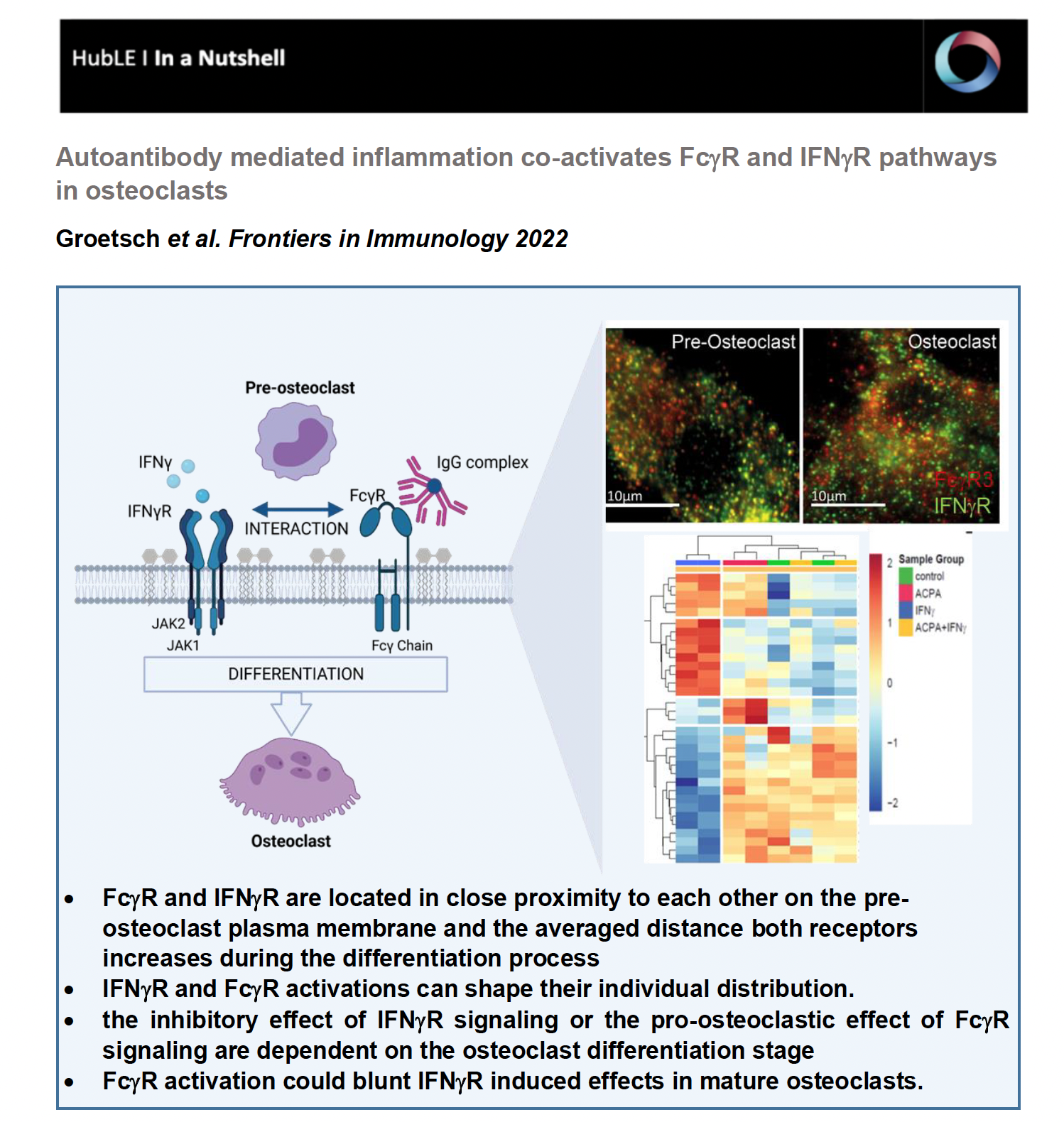
Bone remodeling is a continuous and uninterrupted process in which old bone tissue is removed by osteoclasts (the bone-resorbing cells) while, in parallel, new bone substance is formed by osteoblasts (the bone-building cells). Normal osteoclast activity is essential for the vitality of the tissue and the stability of the bones. In contrast, osteoclasts are responsible for the destruction of bone and cartilage in inflammatory bone diseases such as rheumatoid arthritis (RA).
Rheumatoid arthritis (RA) affects approximately 1% of the Western world population, making it one of the most widespread autoimmune diseases. Inflammatory bone loss in RA is caused by altered bone homeostasis with increased osteoclast generation and activity resulting in accelerated bone resorption. The triggers that lead to progressive bone loss given the appropriate genetic predisposition are still insufficiently researched. It is described that activation of osteoclast differentiation and bone loss in RA is stimulated by IgG immune complexes like anti-citrullinated protein antibodies (ACPAs), which activate the Fc gamma receptor (FcgR) on the osteoclast surface and shift the balance toward bone resorption. Apart from this, by secreting interferon gamma (IFNg), immune cells can block osteoclast differentiation in vitro, although the role of IFNg is discussed controversially. IFNg has been described to induce osteoclast activation in vivo via Tumor necrosis factor alpha (TNFa). Despite the role of IFNg and IgG immune complexes in regulating osteoclast differentiation, no link between these two signaling pathways has been described.
In our study, we hypothesized that bone loss is RA is mutually influenced by the IgG immune complexes formed during the immune response and by the inflammatory cytokine IFNg. In our recently published study in Frontiers in Immunology we aimed to investigate when and how exactly, the FcgR and IFNgR signaling pathways interact with each other to regulate osteoclast differentiation.
Total internal reflection (TIRF) microscopy measurements revealed that FcgR3 and IFNgR are localized in close proximity to each other on the human osteoclast surface. Interestingly, the average distance of FcgR3 and IFNgR increases in the process of osteoclast differentiation, suggesting that the average distance of these receptors is dependent on the maturation state of human osteoclast. Even more, in vitro activation of both receptors indicates that FcgR3 and IFNgR activation influence the membrane localization of the other receptor and possibly shape their individual distribution during activation.
IFNgR activation promotes TRAF6 degradation in vitro, leading to reduced osteoclast formation. However, opposite functions of IFNg have been described depending on the model used. Here we demonstrated that the inhibitory effect of IFNg in vitro depends on the differentiation state of human osteoclasts. While IFNg completely inhibits the differentiation of early osteoclast precursor cells into multinucleated osteoclasts, IFNg promotes fusion of premature osteoclasts. Moreover, in gene expression analyses, IFNgR activation induces FcgR expression in early osteoclasts but not in premature osteoclasts suggesting a co-regulation of FcgR and IFNgR in human osteoclast progenitor cells.
Based on hierarchical cluster analysis of differentially phosphorylated proteins by scioPhospho protein profiling, FcgR and IFNgR activation in premature osteoclasts exhibit distinct clusters compared to non-stimulated controls. Moreover, Volcano plot analyses of the stimulated osteoclasts compared with untreated osteoclasts showed strong induction of protein phosphorylation in IFNg-treated cells but little in ACPA-treated cells. Surprisingly, combined activation of both receptors showed a similar proteomic pattern as the non-stimulated control cells, suggesting a negative balancing mechanism within the two activated pathways. Functional gene ontology (GO) annotation showed an increased phosphoproteome for cellular metabolic (ROS) processes, MAPK signaling, cytoskeleton organization, cell differentiation and activation in IFNg-treated mature osteoclasts that are all in line with the observed stimulatory effect of IFNg on mature osteoclasts. The GO annotation for phosphorylated proteins that were downregulated by IFNg treatment could be clustered to the biological process (BP) of cell differentiation, cell cycle, cell signaling, cell motility, transmembrane transport and migration. In conclusion, the phosphoproteome data show that FcgR or IFNgR activation on multinucleated osteoclasts trigger distinct downstream signaling events while co-stimulated mature osteoclasts clustered close to the control cells. This indicates that FcgR3 activation could attenuate IFNgR induced effects in mature osteoclasts.
In summary, we were able to show for the first time that FcgR3 and IFNgR interact directly with each other during osteoclast differentiation. More specifically, we now know that the IFNgR is dependent on the expression of FcgR3 for efficient signaling; on the other hand, targeted activation of FcgR3 inhibits the inhibitory effect of IFNgR on osteoclast differentiation. Taken together, our data show that FcgR and IFNgR signaling pathways influence osteoclast activity depending on the differentiation state, which may reflect the bone erosion state of rheumatoid arthritis. Our data can be an interesting starting point for the development of new innovative bone building agents and antiresorptive drugs for the treatment of osteoporosis.
These findings are described in the article titled “Inflammatory activation of FcgR and IFNgR pathways co-influence the differentiation and activity of osteoclasts” published in Frontiers in Immunology (2022; doi: 10.3389/fimmu.2022.958974). This work was performed by Bettina Groetsch, Elisabeth Schachtschabel, Philipp Tripal, Benjamin Schmid, Ana-Suncana Smith, Georg Schett and Aline Bozec.